癌症中有数百万种突变和其他遗传变异。然而,与无害的“乘客”相比,了解这些突变中的哪些是有影响的肿瘤“驱动因素”,以及每个驱动因素对癌细胞的作用,一直是一项具有挑战性的工作。许多研究依赖于定制的、耗时的、特定于基因的方法,这些方法为给定突变的更广泛的功能影响提供一维视图。或者,计算预测可以提供功能性见解,但这些发现必须通过实验来证实。
现在,在《自然生物技术》杂志上发表的一份报告中,麻省理工学院和哈佛大学布罗德研究所的一个研究小组公布了一种大规模、高分辨率的方法,用于同时对大量蛋白质编码突变进行功能评估,该方法可以返回丰富的表型信息和这可能被用来研究癌症和其他疾病中任何基因的任何突变。他们通过癌细胞系的概念验证实验获得的结果还表明,个体突变不仅可以对其受影响的基因产生一系列影响还涉及分子途径和整个细胞状态,并为长期接受的将癌症突变分为所谓的“驱动程序”和“乘客”的做法增加了细微差别。
“当你查看来自患者肿瘤的基因数据时,你会发现大多数与癌症相关的突变实际上非常罕见,这意味着我们对这些突变的作用知之甚少,”Broad 癌症项目的 Jesse Boehm 说,他是该研究的共同高级作者,Aviv Regev 是现在在罗氏集团成员基因泰克的广泛核心研究所成员。“要使癌症精准医学成为现实,我们需要对每种突变的功能有深入的了解,但一项重大挑战是定义一种可以在实验室中以所需规模实施的实验方法。这种新方法可能是我们需要的工具。”
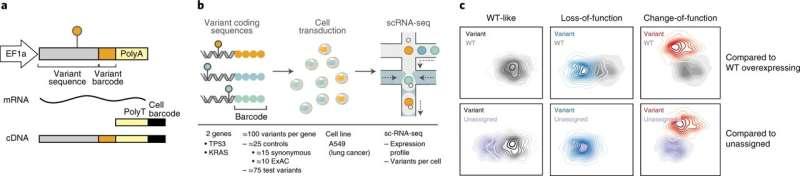
这种称为基于单细胞表达的变异影响表型分析 (sc-eVIP) 的新方法建立在 Perturb-seq 的基础上——一种由 Regev 及其同事在 2016 年开发的方法,用于操纵基因并使用高通量单基因探索这些操纵的后果细胞 RNA 测序和 eVIP,这是 Boehm 及其同事在 2016 年开发的一种方法,用于使用 RNA 测量来分析小规模的癌症变异。虽然 Perturb-seq 分析最初依靠 CRISPR 将突变引入细胞,但 sc-eVIP 团队采用了一种基于过表达的方法,为每个感兴趣的突变设计 DNA 条形码基因构建体,并将它们引入细胞池中,从而细胞以高于正常水平表达突变基因。
然后通过使用单细胞 RNA 测序记录每个受干扰细胞的表达谱,该团队既可以识别给定细胞携带的突变(基于构建体的独特条形码),又可以检查突变对细胞整体表达状态的更广泛影响。这种方法提供了突变对各种分子途径和回路的影响的高度详细的视图,并且不需要针对所研究的每个新基因进行调整。
标签:
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!