新贝叶斯量子算法直接计算原子和分子的能量差
大阪。正如《物理化学化学物理学》杂志最新报道的那样,大阪市立大学科学研究生院的研究人员开发了一种量子算法,可以通过直接计算原子或分子系统的相关状态的能量差来了解它们的电子状态。作为贝叶斯相位差估计实现,该算法打破常规,不关注从相位前和相位后演化计算的总能量差异,而是跟踪能量差异本身的演化。
“几乎所有的化学问题都讨论能量差异,而不是分子本身的总能量,”研究负责人兼特聘讲师 Kenji Sugisaki 说,“此外,出现在元素周期表下部的具有重原子的分子具有大总能量,但化学中讨论的能量差异的大小,例如电子激发态和电离能,与分子的大小无关。”这个想法促使杉崎和他的团队实施了一种直接计算能量差异而不是总能量的量子算法,创造了一个可扩展或实用的量子计算机使我们能够进行实际化学研究和材料开发的未来。
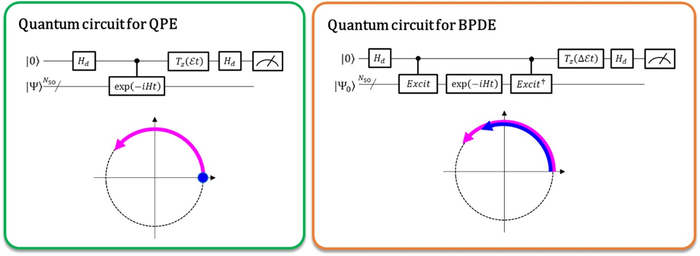
目前,量子计算机能够执行全构型相互作用 (full-CI) 计算,通过称为量子相位估计 (QPE) 的量子算法提供最佳分子能量,并指出对于相当大的分子系统的全 CI 计算是难以处理的。超级计算机。QPE 依赖于这样一个事实:波函数 |Ψ⟩ 表示微观系统的量子态的数学描述——在这种情况下,是微观系统(如原子或分子)的薛定谔方程的数学解——时间——根据它的总能量进化地改变它的阶段。在传统的QPE中,准备了量子叠加态(|0⟩|Ψ⟩+|1⟩|Ψ⟩) ⁄ √2,并且引入受控时间演化算子使得 |Ψ⟩ 仅在第一个量子位指定 |1⟩ 状态时才随时间演化。因此,|1⟩ 状态在时间上创造了进化后的量子阶段,而|0⟩ 状态创造了进化前的量子阶段。进化前和进化后之间的相位差给出了系统的总能量。
大阪市立大学的研究人员将传统的 QPE 推广到直接计算两个相关量子态之间总能量的差异。在新实现的称为贝叶斯相位差估计 (BPDE) 的量子算法中,两个波函数的叠加 (|0⟩|Ψ0⟩ + |1⟩|Ψ1⟩) ⁄ √2,其中 |Ψ0⟩ 和|Ψ1⟩ 分别表示与各态相关的波函数已准备好, |Ψ0⟩ 和 |Ψ1之间的相位差⟩ 叠加后的时间演化直接给出了所涉及的两个波函数之间总能量的差异。“我们强调,该算法遵循能量差异随时间的演变,与单独计算原子或分子的总能量相比,它更不容易产生噪音。因此,该算法适合需要精确能量准确度的化学问题。”国家研究主管和名誉教授Takeji Takui。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!
-
【关于中秋节的英语手抄报】中秋节是中国传统节日之一,每年农历八月十五庆祝。它不仅是家人团聚的日子,也是...浏览全文>>
-
【关于中秋节的小故事】中秋节是中国传统节日之一,承载着丰富的文化内涵和深厚的情感寄托。自古以来,人们在...浏览全文>>
-
【关于中秋节的文化含义】中秋节,是中国传统节日中最具代表性的节日之一,通常在农历八月十五这一天庆祝。它...浏览全文>>
-
【关于中秋节的手抄报内容】中秋节是中国传统节日之一,象征着团圆与丰收。为了帮助同学们更好地了解中秋节的...浏览全文>>
-
【关于中秋节的诗句古诗】中秋节是中国传统节日之一,自古以来便有赏月、吃月饼、家人团聚的习俗。在众多文人...浏览全文>>
-
【关于中秋节的诗歌】中秋节是中国传统节日之一,自古以来就深受文人墨客的喜爱。许多诗人以中秋为题材,创作...浏览全文>>
-
【关于中秋节的诗词精选】中秋节,是中国传统节日中最具诗意的节日之一。自古以来,文人墨客常以中秋为题,写...浏览全文>>
-
【关于中秋节的来历100字】中秋节,又称月圆节,是中国传统节日之一,时间为农历八月十五。其起源与古代对月亮...浏览全文>>
-
【关于中秋节的句子】中秋节是中国传统节日之一,象征着团圆、思念与丰收。自古以来,文人墨客常用诗句表达对...浏览全文>>
-
【关于张起灵身世年龄介绍】在《盗墓笔记》系列小说中,张起灵是一个神秘而重要的角色,他的身世和年龄一直是...浏览全文>>